FusionCAT es un proyecto que tiene como objetivo promover la colaboración entre universidades y centros de investigación de Cataluña mediante el establecimiento de una comunidad de investigación activa hacia el desarrollo de tecnologías de energía de fusión. El proyecto está coordinado por el Barcelona Supercomputing Center – Centro Nacional de Supercomputación (BSC, https://www.bsc.es) que participa en todos los grupos de trabajo técnico. La fabricación de materiales seguros y duraderos para los reactores de fusión es un avance fundamental hacia la energía de fusión. El estudio y desarrollo de materiales para reactores de fusión (paquete de trabajo 3.4 del proyecto) es una de las tecnologías clave a desarrollar durante este proyecto, y es llevada a cabo por el BSC Fusion Group. En particular, los investigadores del BSC Julio Gutiérrez Moreno, Stephan Mohr y Mervi Mantsinen lideran las tareas de FusionCAT en simulaciones atomísticas de materiales basadas en la teoría del funcional de la densidad (DFT) con el código BigDFT.
Durante los últimos años, el tungsteno (W) se ha convertido en un uno de los principales candidatos para material de primera pared en reactores de fusión gracias a su resistencia, alto punto de fusión y baja retención de tritio. Además, el tungsteno sus compuestos se utilizan en diversas aplicaciones tecnológicas, como por ejemplo en catálisis o nanoelectrónica. La estabilidad y propiedades de los materiales está directamente ligada su configuración electrónica, de este modo, el estudio de la estructura electrónica del W es clave para el diseño e implementación de tecnologías basadas en este material. Sin embargo, y a pesar de los grandes esfuerzos llevados a cabo en décadas recientes, la estructural electrónica del W no ha sido caracterizada a forma precisa aún a día de hoy.
En un artículo reciente publicado en Physical Review B titulado Lifetime effects and satellites in the photoelectron spectrum of tungsten metal (ver artículo aquí), se han combinado experimentos de última generación y métodos teóricos de cálculo ab-initio para proporcionar probablemente el análisis más preciso de la estructura electrónica del tungsteno hasta la fecha.
Este trabajo ha sido dirigido por Curran Kalha y Anna Regoutz (grupo de Espectroscopía de Rayos-X Aplicada – Applied X-ray Spectroscopy (AXS) group en la University College London), quienes caracterizaron el espectro de tungsteno utilizando una combinación de espectroscopia de fotoelectrones de rayos X duros y blandos de alta resolución (SXPS y HAXPES) con espectroscopía de pérdida de energía de electrones en el modo de reflexión (REELS).
Las mediciones experimentales han sido comparadas con cálculos ab-initio, que respaldan la interpretación de los complejos espectros de tungsteno. Los cálculos ab-initio con BigDFT se han utilizado para calcular la densidad proyectada de estados (PDOS) del tungsteno. El trabajo computacional ha sido realizado por el equipo de modelado de materiales del BSC para FusionCAT en estrecha colaboración con la desarrolladora de BigDFT Laura Ratcliff (Imperial College London). BigDFT es un código preciso y flexible que realiza simulaciones ab-initio a escala atómica. El código se desarrolla conjuntamente en BSC y es una de las herramientas clave utilizadas en las actividades de modelado de materiales del proyecto FusionCAT. La versión de escala lineal del código (LS por sus siglas en inglés) puede modelar sistemas atómicos con miles de átomos con alta precisión y reproducibilidad, superando las limitaciones de escalado del DFT tradicional, donde el coste computacional aumenta cúbicamente con tamaño del sistema. Los cálculos de LS-BigDFT (presentados en la Figura 1.a) coinciden con los resultados de otros métodos teóricos utilizados en este estudio y son comparables con las mediciones experimentales en la banda de valencia, identificando claramente el espectro y posiciones relativas de energía. Estos resultados sientan las bases para describir sistemas más complejos como estructuras de tungsteno desordenadas, defectuosas o dopadas en estudios futuros con alta precisión y reproducibilidad.
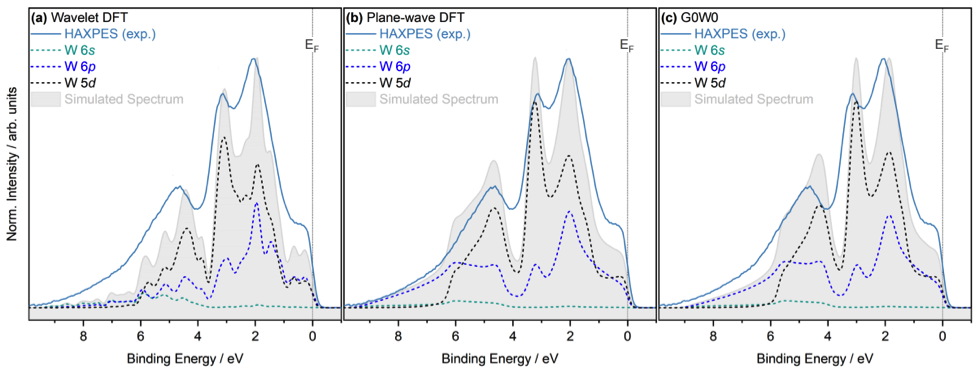
Figura 1. Comparación de los espectros de densidad proyectada de estados proyectados (PDOS) calculados con DFT y G0W0 con los espectros de banda de valencia de la espectroscopia de fotoelectrones de rayos X duros (HAXPES). (a) Cálculos de LS-BigDFT utilizando un conjunto de funciones de base de ondículas (wavelets), (b) DFT con Quantum Espresso utilizando ondas planas y (c) cálculos de G0W0. Figura adaptada de arXiv:2109.04761. Más detalles sobre las medidas experimentales y métodos teóricos, y una descripción más detallada del trabajo se pueden consultar en las siguientes versiones del artículo aquí o aquí
Además, el análisis exhaustivo del espectro del núcleo, obtenido por la combinación de mediciones con SXPS y HAXPES, proporciona nueva información sobre la naturaleza de las transiciones específicas que explican los picos satélite observados. Las funciones espectrales calculadas a partir de la teoría de perturbaciones de muchos cuerpos (many-body perturbation theory o MBPT) basada en método de función e Green GW y «GW más cumulante» (GW+C) se han utilizado para respaldar las asignaciones experimentales del espectro. Estos cálculos han sido llevado a cabo por el grupo de investigación liderado por Johaness Lischner en el Imperial College London.
Los resultados y la metodología detallados en este artículo proveen de información clave para las aplicaciones fundamentales e industriales del tungsteno y serán útiles para la exploración futura de otros materiales con estructuras electrónicas complejas.
- El artículo puede ser consultado en web de la revista: https://journals.aps.org/prb/abstract/10.1103/PhysRevB.105.045129
- La versión inicial (preprint) se puede descargar en arXiv:2109.04761v1
- Todos los espectros de SXPS y HAXPES están disponibles en: https://doi.org/10.6084/m9.figshare.16432617
- Todos los datos y archivos utilizados para los cálculos con LS-BigDFT están disponibles en el repositorio NOMAD: https://dx.doi.org/10.17172/NOMAD/2021.08.27-1
Los autores agradecemos el acceso a recursos computacionales y soporte técnico en MareNostrum proporcionado por BSC (RES-QS-2020-3-0026) y la colaboración EU-JA Broader Approach en el Computational Simulation Center of International Fusion Energy Research Center (IFERC-CSC) . Este trabajo ha sido financiado por el proyecto FusionCAT con número de referencia 001-P-001722, cofinanciado por el Fondo de Desarrollo Regional de la Unión Europea en el marco del Programa Operativo FEDER de Cataluña 2014-2020, con el apoyo de la Generalitat de Cataluña.