FusionCAT is a project that aims to promote collaboration between universities and research centres in Catalonia by establishing an active research community towards the development of fusion power technologies. The project is coordinated by the Barcelona Supercomputing Centre – Centro Nacional de Supercomputación (BSC, https://www.bsc.es) that participates in all the technical work packages. Manufacturing safe and durable materials for fusion reactors is critical advancement towards fusion power. The study and development of materials for fusion reactors (project’s work package 3.4) is one of the key technologies to develop during this project, and it is carried out at the BSC Fusion Group. In particular, the BSC researchers Julio Gutiérrez Moreno, Stephan Mohr and Mervi Mantsinen led the FusionCAT tasks on atomistic simulations of materials based on ab-initio density functional theory (DFT) calculations with the BigDFT code.
During the past few years, tungsten (W) has emerged as a promising candidate for plasma-facing applications in fusion reactors thanks to its strength, high melting point and low tritium retention. Moreover, tungsten and tungsten-based compounds are used in various technological applications, such as catalysis or nanoelectronics. The investigation of the electronic structure is highly relevant today, and it is key for the design and implementation of tungsten-based technologies, as this is strongly related to the stability and properties of the material. However, despite the large efforts in studying the electronic properties of tungsten metal using both theoretical and experimental approaches, some complex features are still not properly characterized.
In a recent article published in Physical Review B and entitled Lifetime effects and satellites in the photoelectron spectrum of tungsten metal (see article here), state-of-the-art experiments and ab-initio theoretical approaches have been combined to provide the most accurate description of the electronic structure of tungsten to date. This work has been led by Curran Kalha and Anna Regoutz (Applied X-ray Spectroscopy (AXS) group at University College London), who characterised the tungsten spectrum using a combination of high-resolution soft and hard x-ray photoelectron spectroscopy (SXPS and HAXPES) with reflection electron energy loss spectroscopy (REELS).
Experimental measurements are compared with ab-initio calculations, which support the interpretation of the complex tungsten spectra. In particular, BigDFT calculations have been used to calculate tungsten’s projected density of states (PDOS). This work has been performed by the BSC FusionCAT materials modelling team in close collaboration with the BigDFT developer Laura Ratcliff (Imperial College London). BigDFT is a precise and flexible code that performs ab-initio simulations at the atomic scale. The code is jointly developed at BSC and is one of the key tools used in FusionCAT materials modelling activities. The Linear-Scaling (LS) version of the code can model atomic systems with thousands of atoms with high accuracy and reproducibility, overcoming the computational cost limitations of traditional cubic-scaling DFT. LS-BigDFT calculations (presented in Figure 1.a) agree very well with the other theoretical methods used in this study and are comparable with the experimental measurements at the valence band, clearly identifying all key features and their relative energy positions. These results settle the base for describing more complex systems such as disordered, defective or doped tungsten structures in future studies with high accuracy and reproducibility.
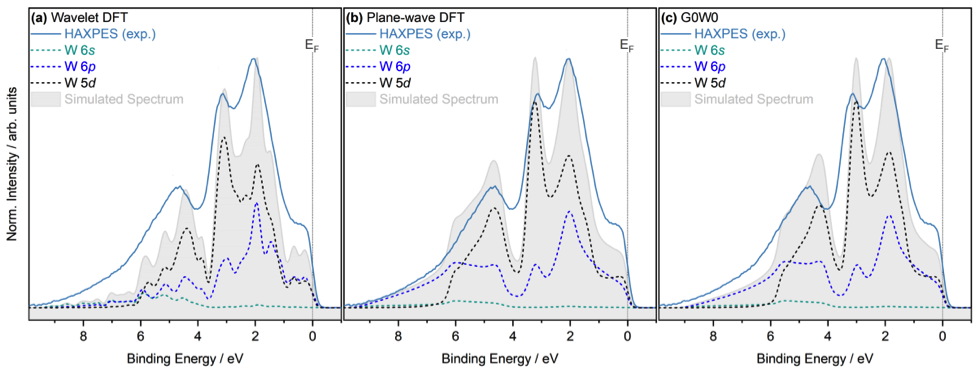
Figure 1. Comparison of simulated projected density of states (PDOS) spectra calculated using density functional theory (DFT) and G0W0 approaches with the hard x-ray photoelectron spectroscopy (HAXPES) valence band spectra. (a) LS-BigDFT calculations using a wavelet basis set, (b) DFT with Quantum Espresso using a plane-wave basis set, and (c) G0W0 calculations. Figure adapted from arXiv:2109.04761, more details on the experimental and theoretical setups and description can be found here or here
Moreover, this work’s exhaustive analysis of SXPS and HAXPES core-level spectra provides new insights into the nature of specific transitions underlying the observed satellite features. Spectral functions calculated from Many-Body Perturbation Theory (MBPT) within the GW and “GW plus cumulant” (GW+C) approaches have been used to support the experimental assignments. These calculations have been carried out by Johaness Lischner’s team at Imperial College London.
The results and methodology detailed in this paper provide key insights for fundamental and industrial applications of tungsten and will be of use for the future exploration of other materials with complex electronic structures.
- The article can be accessed at the publisher website: https://journals.aps.org/prb/abstract/10.1103/PhysRevB.105.045129
- The initial preprint version can be downloaded from arXiv:2109.04761v1
- All survey, core level and valence band spectra from SXPS and HAXPES are freely available at: https://doi.org/10.6084/m9.figshare.16432617
- Datasets from LS-BigDFT calculations are also openly accessible at the NOMAD repository: https://dx.doi.org/10.17172/NOMAD/2021.08.27-1
We acknowledge the access to computational resources and technical support at MareNostrum provided by BSC (RES-QS-2020-3-0026) and the EU-JA Broader Approach collaboration in the Computational Simulation Centre of International Fusion Energy Research Centre (IFERC-CSC). This work was financially supported by the FusionCAT project with reference number 001-P-001722, co-financed by the European Union Regional Development Fund within the framework of the ERDF Operational Program of Catalonia 2014-2020, with the support of Generalitat of Catalonia.